Where Compassion Inspires Progress

Dr. Flanagan-Steet’s research has largely focused on defining the mechanisms governing early tissue development. This has ranged from investigating how neuromuscular synapses form to development of the embryonic heart and craniofacial skeleton. For nearly twenty years Dr. Flanagan-Steet’s efforts have centered on defining the molecular and cellular mechanisms underlying pathogenesis of rare genetic diseases. This has included several lysosomal storage disorders (LSDs) as well as the congenital disorders of glycosylation (CDG). Her work on genetic diseases largely involves generating zebrafish models to investigate gene function and disease pathogenesis. This work pioneered the use of zebrafish to model rare inherited diseases, bringing new insight into the molecular initiators and mechanisms underlying pathogenesis of mucolipidosis II and PMM2-CDG. In her role as Director of Functional Studies, she works closely with the Divisions across the Center to advance our understanding of rare diseases. The team uses a combination of genetic, molecular, biochemical and microscopic approaches to answer questions of disease pathogenesis. We have adapted both traditional and modern biochemical procedures to extend the utility of our zebrafish models to functionally understand the proteins and genes we are studying. We are keenly interested in elucidated the full pathogenic cascade so that we may identify biochemical pathways downstream of the causative genetic mutation that may be modulated for disease therapy. A summary of current projects is shown below:
Disease models for the Congenital Disorders of Glycosylation (CDG). We are using zebrafish models of PMM2-CDG to identify pathogenic cascades driving cartilage and bone malformations, as well as neuromuscular issues. We are currently addressing whether altered processing of specific proteins may initiate disease pathogenesis in these tissues. Our group is also interested in defining how loss of subunits of the OST complex impact development. We are using TALENS and CRISPR-Cas9 tools to generate zebrafish models of newly identified CDGs. Novel CDG models were initiated using patient-derived insight regarding CDG-associated genes.
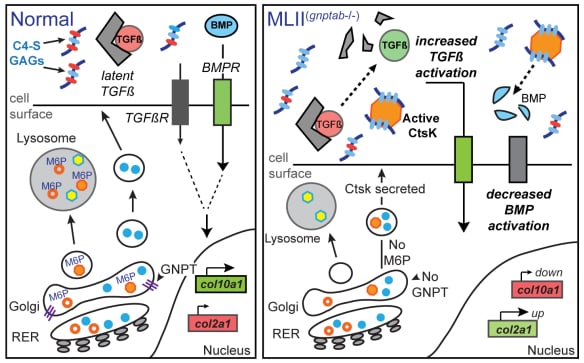
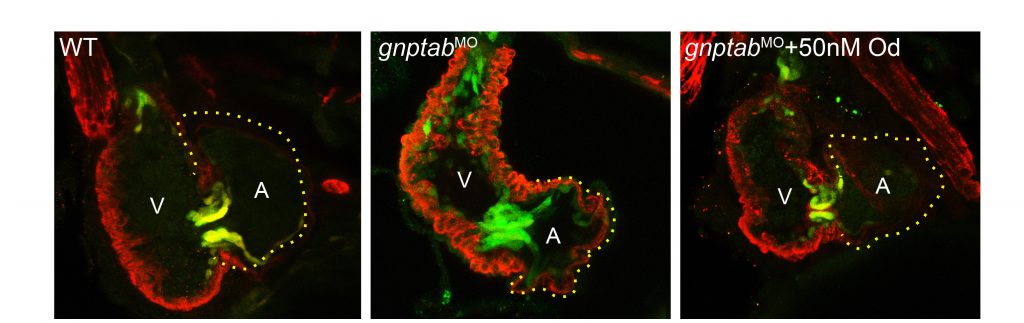
Defining the pathogenesis of lysosomal storage disorders. Our group has been interested in understanding the pathogenic mechanisms associated with loss of carbohydrate-dependent lysosomal sorting for many years. Using several zebrafish models of mucolipidosis II (MLII, I-cell), we have identified cathepsin proteases as key pathogenic initiators. When mislocalized outside the cell these proteases can inappropriately act on numerous substrates, including growth factors such as TGFß, causing abnormal chondrogenesis (see Figure 1). We continue to explore how extracellular cathepsin proteases influence normal and disease tissue development. Our work has largely focused on abnormal cartilage development, but recent efforts include the congenital heart defects associated with MLII. Recent efforts have extended this work to include other LSDs, exploring the extent to which similar mechanisms drive disease in MLII-related disorders.

©2024 Greenwood Genetic Center. All Rights Reserved. Privacy Policy. Notice of Nondiscrimination. Good Faith Estimate. Code of Conduct. Sitemap.
